Meta analysis of gene expression data of multiple cancer types to predict biomarkers and drug targets 
2 Department of Biological sciences, Scientific Bio-Minds, Bangalore, India
 Author
Author  Correspondence author
Correspondence author
Computational Molecular Biology, 2015, Vol. 5, No. 5 doi: 10.5376/cmb.2015.05.0005
Received: 17 Aug., 2015 Accepted: 25 Sep., 2015 Published: 19 Oct., 2015
Shashank K.S., Mamatha H.R., and Prashantha C.N., 2015, Meta Analysis of Gene Expression Data of Multiple Cancer Types To Predict Biomarkers and Drug Targets Interactions in Ovarian Cancer, Computational Molecular Biology, 5(5): 1-9
Meta analysis of gene expression data of multiple cancer types such as breast, colon and ovary used to identify gene signatures that functionally used as a marker to prognosis and molecular diagnostics. There is a reliable identification of gene signatures is associated with different cancer types remains a challenge. The aim of this study is to develop microarray statistical data analysis methods and SVM classifiers to identify differentially expressed genes in different cancer types. Using our method to perform 16 datasets such as 6 breast cancer, 4 colon cancer and 6 ovarian cancer of different datasets. Our results is analysed in 4 different methods (a) preprocess the data to identify quality expression of datasets by removing null values and non significant values (p<0.05) (b). Differential gene expression analysis using statistical analysis to predict upregulation and downregulated gene signatures (c) subgrouping of datasets that has been classified based on cancer types (d) gene network prediction to identify gene-gene interaction to understand biological markers. We have predicted 8 markers in breast cancer, 10 markers in colon cancer and 16 markers in ovarian cancer is providing new direction for diagnostics and therapeutic development.
Introduction
Cancer is a large family of disease that can threat to people’s health ad life. According to 2014-15 survey shows 22% of disease death is observed in worldwide (Cancer Fact sheet N°297, WHO, 2014). In India cancer is second largest death following with heart disease. The statistical survey of cancer shows 82% of women is affecting breast cancer, 62% of men and women is affecting with colon and 90% of women is affecting with ovarian cancer (Matsushita K et al.,2010) The decreased trend in diagnostics techniques for identification of cancer in early stage of development. In the 21st decade, molecular biomarkers that helps to identify disease in early stage. In the current research using three cancer types (breast, colon and ovarian cancer) is helps to identify molecular markers using microarray technique.
In the present year 9.1 million women is affecting with breast cancer in worldwide. In addition 232,670 women is diagnosed with in a year. 30% of women population is affected due to genetic abnormalities such as mutation of BRCA1 ad BRCA2 genes (Dumitrescu RG et al.,2005). In addition there are some other oncogenic genes such as k-RAS, p53, PTEN, NBS1 etc also causing breast cancer (Honrado E et al.,2006). The colon cancer also leading cancer types that frequently affecting other tissues such as lungs, breast and prostrate tissues. The genetic alterations of k-RAS, APC, P53, β-catenin, GSK-3β that mainly affect WNT- β-catenin signaling pathways that also affects breast and ovarian cancer (Vogelstein B et al., 1988), (Fearon ER et al., 1990). The epithelial ovarian cancer is also dangerous cancer type in women, the mutation of p53, BCL-XL, EGFR, MDM2, MCI-2, NOXA etc is mainly involved in ovarian cancer (Baekelandt M et al., 1999), (Kupryjanczyk J et al., 2003), (Nielsen JS et al., 2004). A number of genetic marker has been proposed to identify cancer such as BRCA1, BRCA2 of breast cancer, APC, GSK-3β of colon cancer and CA125 of ovarian cancer. In addition there are more number of serum markers that helps to clinically diagnostics through breast, colon and ovarian cancer. However, its effectiveness of detecting more genetic markers that widely considered to use more advanced techniques such as DNA microarray technology to identify genetic profiling of cancer by allowing thousands of genes that significantly associated with cancer types (Golub TR 2001), (Elvidge G 2006).
Herein we present computational methods to predict genetic markers of three types of cancer based on microarray data. Using different biological algorithms to preprocess the data that significantly predicts the quality of intensity values that shows screening of datasets. Using statistical techniques to predict differential gene expression of both upregulated and down regulated. We have compared the Meta analysis to predict markers of multi-cancer types are based on high level computing power. Using gene-gene network study shows various significant genes that specifically regulated either with multi cancer types specifically. We believed to these novel genes that shows gene profiling expression will provide high valuable markers those new approaches in diagnostics and therapeutics.
Materials and Methods
The Meta analysis of three different cancer diseases such as breast, colon and ovarian cancer datasets were retrieved from GEO database. The Datasets of breast cancer (GEO ID: GSE30543) of 6 samples with SUM149 control siRNAs and siRNA targeting TIG1 replicates (Wang X1 et al., 2013). The colon cancer dataset (GSE34299) of 4 samples has HT29 parental cell lines and HT29RC PLX4720 resistant cell lines grown in increasing concentration of the drug to develop acquired resistance (Mao M1 et al., 2012). The ovarian cancer dataset (GSE35972) of 6 samples has untreated TOV112D cells and NSC319726 treated with different biological replicates (Yu X1 et al.,2012). All the datasets with different samples is analyzed using GPL570 (HG-U133_Plus_2) Affymetrix human genome array platform. All probe sets of HG-U95Av2 is identically replicated on the diseased transcript variant. The RNA probe sets were derived from RefSeq, dbEST and GenBank. The sequence clusters were created from the UniGene database and gene names were refined by publicly available databases. Using Statistical analysis software such as R and BioConductor to analyze pre-processing and differential gene expression to classify breast, ovarian and colon cancer genes that used as a potential drug targets.
Pre-processing of Raw Microarray data
Using Affy and affycoretools of BioConductor packages is used to pre-process the data (R Core Team 2012), (Robinson MD et al., 2010). There are different algorithms is used such as RMA and MAS5 algorithms that helps to pre-process the data to correct both foreground and background intensities of all probes. Different statistical techniques used in normalization of probe sets such as constant, quintiles and invariant set that predicts the PM and MM corrections. However, the signal intensity for MM probe can often be larger than PM probe implying that MM probe is detecting true signal as well as background signal. After correction of all intensity levels is used for differential gene expression in each disease datasets.
Differential gene Expression analysis
After pre-processing of datasets, the resultant CEL files used for differential gene expression. Using Limma packages to predict differential gene expression data arising from microarray RNA samples. For datasets of control and cancer samples of differential levels change between two samples which genes are up-regulated (increased in expression) or down-regulated (decreased in expression). The clustering of genes that follows expression patterns across a set of samples, or clustering samples with similar expression patterns across genes. Each sample group will contain numerous replicates. The group expression level for a probe will be summarized as the mean of the expression levels in the group replicates. Thus, differential expression problems are a comparison of means. When there are two sample groups this is a t test of some kind (Prashantha N et al., 2013). The clustering of up regulated and down regulated genes were predicted with clustering experiment. Hierarchical clustering of differentially expressed genes with respect to probable expression of B values with correlation coefficient of control-cancer datasets. The relationship among objects are represented by a tree whose branch lengths id different with differential gene expression (Parker JS et al.,2009). The differential expressed genes were annotated using GO.db package. The Annotation of HGU 133plus 2 package of GO annotation helps to understand the genes involved in differential expressed genes along with biological process, molecular function or cellular components of genes with systematic classification.
Comparative analysis of differentially expressed genes
Using geNETClassifier algorithm to classify the genes was differentially expressed in different disease datasets along with gene networks. The genome-wide association studies of expression sets or expression matrix files of ranked genes, probe sets of different variables is optimized with training sets. Using multi-class SVM based classifier to quires genes chosen for classification; the mutual-information (interactions) and the co-expression (correlations) between the genes are also calculated and analyzed by the algorithm. These allow estimating the degree of association between the variables and they are used to generate a gene network for each class. These networks can be plotted, providing an integrated overview of the genes that characterized each disease (i.e. each class).
Functional Annotation and Enrichment Analysis
In order to obtain the functional enrichment of the differentially expressed genes on the cell level, we used the GO (Gene Ontology) database to classify the gene function and location information. We performed GO cluster analysis by using the cluster Profiler package, then deduced the affection of these differentially expressed genes to the cells by cluster the cells within the molecular functions and biological processes. The Database for Annotation, Visualization and Integrated Discovery (DAVID) (http://david.abcc.ncifcrf.gov) and GOrilla tool were used to identify over-represented biological functions and pathways among the differentially expressed genes.
Results and Discussion
This study is focused on three of the most prevalent cancer types such as 6 breast cancer, 4 colon and 6 ovarian cancer microarray datasets is available in publicly available GEO database. The datasets contains cancerous genome sets corresponding with control tissues that help to predict drug targets of each individual cancer types or groups of cancers as gene-gene interactions.
Prediction of drug targets for individual cancer types
We have searched individual datasets of each cancer types whose gene expression patters is classified based on cancer types and control tissues. Specifically, all datasets is classified to predict drug targets that specifically distinguishing the cancer types of both disease and control types. In addition, we have ranked the k-genes that significantly expressed in both upregulated and down regulation that classified based on gene-gene interaction studies.
A. Breast cancer
The analysis of breast cancer dataset contains 6 samples such as 3 SUM149 cells transfected with control sample of siRNA and SUM149 cells transfected with siRNA targeting of tarzarotene-induced gene 1 (TIG1). All these 6 samples is annotated with hgu133plus2 contains 54675 genes, using normalization methods to filter the genes that significantly associated with p-values, we have filter the 54675 genes of which 12788 genes that has significantly associated with gene expression. 1220 genes have upregulated and 11568 genes are downregulated that differentially expressed in breast cancer. Using SVM classification, we have identified the 1275 most common significant genes that associated with breast cancer. Among the 1275 genes 751 genes that encodes proteins, these protein codes genes that helps to predict disease target genes that helps for drug targets to control disease (Figure:1). Using functional annotation and enrichment analysis 130 genes that significantly upregulated these genes such as SHISA2, FBXO23, mmp7, fn1, Cfi, Egr1, DCLK1, DCN, SERPINB3, SERPINB4, MAP3K4, ITGBL1, OLFML3, NPY1R and PHLDA1 genes is mainly associated with transcriptional regulation of breast cancer (table:1). Using gene-gene interaction studies of both classes shows 30 genes that significantly associated with gene regulation. There are 8 genes such as FBXO23, MMP7, FN1, CFI, DCN, SERPINB3, SERPINB4 and MAP3K4 is expressed in blood serum within breast cells and is used for potential serum biomarkers (table:1, 2) (Figure: 2).
|
|
|
|
|
|
|
|
.png)
B. Colon Cancer
Our analysis was done using 4 datasets of which 2 samples are HT29 parental cell lines and another two samples are HT29RC PLX4720 resistant cell lines. We have found 268 upregulated genes is consistently expressed in HT29RC cell lines. There are 1268 down regulated genes also associated with both cell lines and control tissues. Using SVM classifier to differentia more significant genes of both conditions that differentially expressed in colon cancer has 60 genes, out of 60, 45 genes has protein expression these genes is combines together with both upregulated and down regulated within colon tissues (Figure: 3). We have found 10 genes is generally expressed on blood serum of colon tissues and is potentially used as a best predicted biomarkers. Using functional annotation and enrichment analysis of differentially expressed genes shows that KRAS, DPT, PROM1, MMP1, MMP7, FBN2, MAOB, SPRR3, PHLDB2, EMP1, DCLK1, AKAP12 genes that regulate colon cancer (table: 3, 4).
The MMP1 and MMP7 genes is mainly involved in colon tissues by regulating immune response, K-ras are an oncogene it helps to transcriptional regulation by inhibiting p53 gene expression with MDM2 receptor to cause colon cancer (Figure: 4).
|
|
|
|
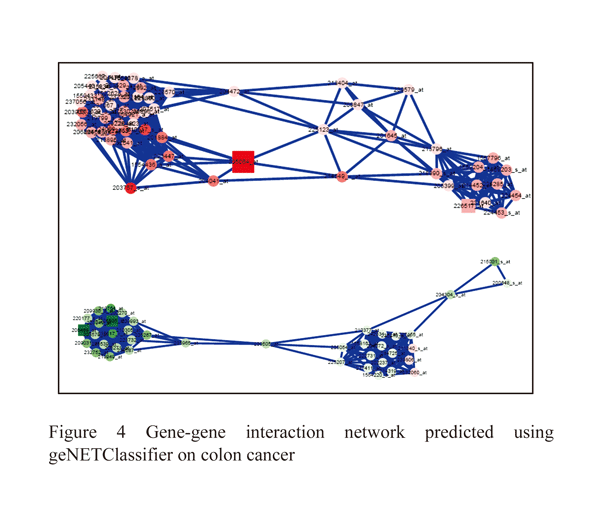
|
|
|
|
C. Ovarian cancer
The ovarian cancer studies has 6 samples of which 3 samples is p53 targeted TOV112D cells untreated and other 3 samples P53 targeted NSC319726 cells data. There are total of 54675 genes of which 1566 genes is significantly associated differential expression in ovarian cancer. There are 810 genes is upregulated within ovarian tissues of which p53 associated with cancer and control tissues, 756 genes dow regulated based on significant test (Figure: 5). Using SVM classification methods we have classified both significant genes in both the datasets shows only 309 genes is probabilistic expression with ovarian cancer. 175 genes is p53 mutation association with TOV112D and NSC319726 cells, these genes is targeted for ovarian cancer therapeutic properties and is used for best biomarkers (table:5,6). The significant genes associated with ovarian cancer such as CDKN1A, PTEN, MDM2, DDB2, GADD45A, FANCC, HRAS, MLH1, DNMT1, VDR, PMS2, APC, TP53I3, MSH2, IGFBP3, EGFR, APC, MSH2, MET, CHMP4C, BIRC5, EGFR, TP53 TP63 (Figure: 6).
|
|
|
|
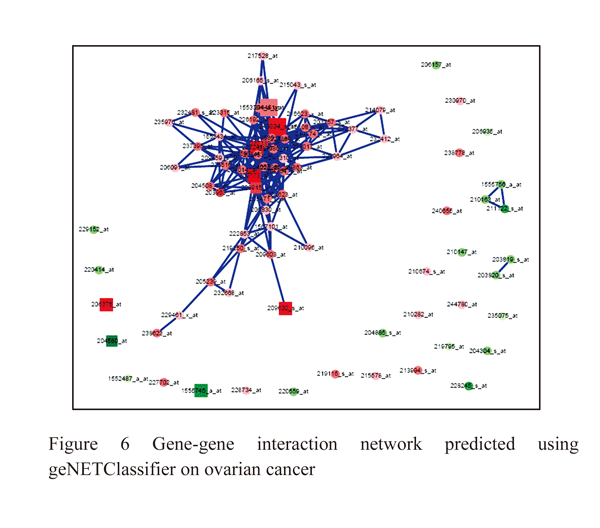
|
|
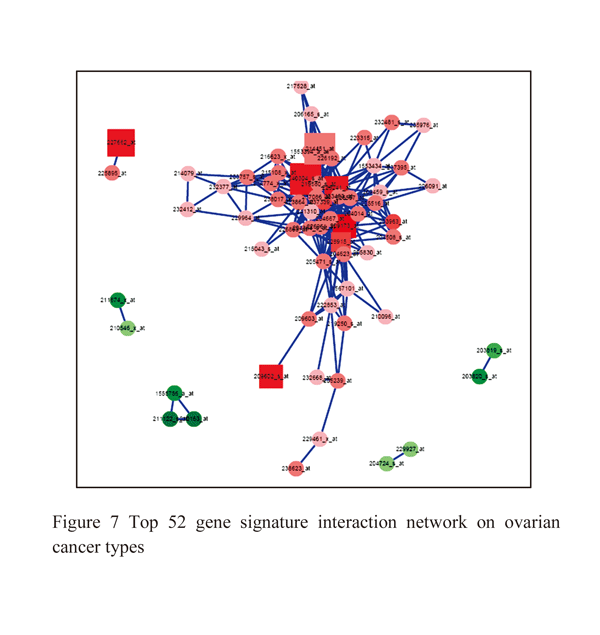
|
|
.png)
|
|
.png)
Conclusion
A statistical methods to predict differential gene expression analysis that significantly associated with cancer and control samples of breast, colo and ovarian cancer cell types. A different computational protocols for predicting biomarkers in cancer tissues that corresponds with individual gene markers. Using functional enrichment analysis across all cancer types have identified different functional characters of genes that specifically helps for biomarkers. Using this application is helps to identify biomarkers for further diagnostics to identify disease in early stage of infection and disease progression. The information provided on individual genes that should provide useful information to elucidating pathways in cancer as well as expeditig the search for potential drug targets for specific cancer.
Acknowledgement
We are thankful to Prashantha CN, founder CEO and Managing Director, Scientific Bio-Minds, Bangalore to supporting laboratory facility to design the project, objectives, practical methods and manuscript preparation. We are also thankful to Mamatha HR Department of Computer Science to verify the practical methods are step-by-step and providing valuable guidance to make my objectives successful. We also thank to my parents to providing valuable support to continue my project successful. We are more grateful to Prashantha CN for successfully guiding project.
Conflict of interest
All authors are accepted for publishing this article. No authors will conflict this article.
References
Baekelandt M, Kristensen GB, Nesland JM, Tropé CG, Holm R. 1999. “Clinical significance of apoptosis-related factors p53, Mdm2, and Bcl-2 in advanced ovarian cancer.” J Clin Oncol, 17: 2061
Cancer Fact sheet N°297". Feb 2014. “World Health Organization”. Retrieved 10 June 2014.
Dumitrescu RG, Cotarla I.2005. “Understanding breast cancer risk--where do we stand in 2005?” J Cell Mol Med, 9:208–21
http://dx.doi.org/10.1111/j.1582-4934.2005.tb00350.x
Elvidge G. 2006. “Microarray expression technology: from start to finish.” Pharmacogenomics 7: 123–134.
http://dx.doi.org/10.2217/14622416.7.1.123
Fearon ER and Vogelstein B. 1990. “A genetic model for colorectal tumorigenesis.” Cell, 61, 759–767.
http://dx.doi.org/10.1016/0092-8674(90)90186-I
Golub TR. 2001. “Genome-wide views of cancer.” N Engl J Med 344: 601–602
http://dx.doi.org/10.1056/NEJM200102223440809
Honrado E, Osorio A, Palacios J, Benitez J. 2006. “Pathology and gene expression of hereditary breast tumors associated with BRCA1, BRCA2 and CHEK2 gene mutations.” Oncogene, 25:5837–45
http://dx.doi.org/10.1038/sj.onc.1209875
Kupryjańczyk J, Szymańska T, Madry R, Timorek A, Stelmachów J, Karpińska G, Rembiszewska A, Ziółkowska I, Kraszewska E, Debniak J, Emerich J, Ułańska M, Płuzańska A, Jedryka M, Goluda M, Chudecka-Głaz A, Rzepka-Górska I, Klimek M, Urbański K, Breborowicz J, Zieliński J, Markowska J. 2003. “Evaluation of clinical significance of TP53, BCL-2, BAX and MEK1 expression in 229 ovarian carcinomas treated with platinum-based regimen.” Br J Cancer, 88: 848–854
http://dx.doi.org/10.1038/sj.bjc.6600789
Matsushita K, van der Velde M, Astor BC, Woodward M, Levey AS, de Jong PE, Coresh J, Gansevoort RT. Chronic Kidney Disease Prognosis Consortium. 2010. “Association of estimated glomerular filtration rate and albuminuria with all-cause and cardiovascular mortality in general population cohorts: a collaborative meta-analysis.” Lancet, 375:2073–2081.
http://dx.doi.org/10.1016/S0140-6736(10)60674-5
Mao M, Tian F, Mariadason JM, Tsao CC, Lemos R Jr, Dayyani F, Gopal YN, Jiang ZQ, Wistuba II, Tang XM, Bornman WG, Bollag G, Mills GB, Powis G, Desai J, Gallick GE, Davies MA, Kopetz S. 2012. “Resistance to BRAF inhibition in BRAF-mutant colon cancer can be overcome with PI3K inhibition or demethylating agents.” Clin Cancer Res, 19(3):657-67.
Nielsen JS, Jakobsen E, Hølund B, Bertelsen K, Jakobsen A. 2004. “Prognostic significance of p53, Her-2, and EGFR overexpression in borderline and epithelial ovarian cancer.” Int J Gynecol Cancer 14: 1086–1096
http://dx.doi.org/10.1111/j.1048-891X.2004.14606.x
Prashantha Nagaraja, Kavya Parashivamurthy, Nandini Sidnal, Siddappa Mali, Dakshyani Nagaraja, and Sivarami Reddy. 2013, “Analysis of gene expression on ngn3 gene signaling pathway in endocrine pancreatic cancer.” Bioinformation, 9(14): 739–747.
http://dx.doi.org/10.6026/97320630009739
Parker JS, Mullins M, Cheang MCU, Leung S, Voduc D. 2009. “Supervised Risk Predictor of Breast Cancer Based on Intrinsic Subtypes.” Journal of Clinical Oncology 27: 1160–1167.
http://dx.doi.org/10.1200/JCO.2008.18.1370
R Core Team, 2012. “R: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing.” http://www.R-project.org/.
Robinson MD, McCarthy DJ, Smyth GK. 2010. “EdgeR: a bioconductor package for differential expression analysis of digital gene expression data.” Bioinformatics, 26:139–140.
http://dx.doi.org/10.1093/bioinformatics/btp616
Vogelstein B, Fearon ER, Hamilton SR, Kern SE, Preisinger AC, Leppert M, Nakamura Y, White R, Smits AM and Bos JL. 1988. “Genetic alterations during colorectal-tumor development.” N. Engl. J. Med., 319, 525–532.
http://dx.doi.org/10.1056/NEJM198809013190901
Wang X1, Saso H, Iwamoto T, Xia W, Gong Y, Pusztai L, Woodward WA, Reuben JM, Warner SL, Bearss DJ, Hortobagyi GN, Hung MC, Ueno NT. 2013. “TIG1 promotes the development and progression of inflammatory breast cancer through activation of Axl kinase.” Cancer Res, 73(21):6516-25.
http://dx.doi.org/10.1158/0008-5472.CAN-13-0967
Yu X, Vazquez A, Levine AJ, Carpizo DR. 2012. “Allele-specific p53 mutant reactivation.” Cancer Cell, 21(5):614-25.
http://dx.doi.org/10.1016/j.ccr.2012.03.042



. PDF(688KB)
. FPDF(win)
. HTML
. Online fPDF
Associated material
. Readers' comments
Other articles by authors
. Shashank K.S
. Mamatha H.R
. Prashantha C.N
Related articles
. breast cancer
. Colon cancer
. Ovarian cancer
. Microarray
. Statistics
. Limma
. Biocoductor
. geNETClassifier
Tools
. Email to a friend
. Post a comment